Mechanism of action.
Pipeline.

Our clinical pipeline centers on orphan kidney diseases of high unmet need, including Autosomal dominant polycystic kidney disease (ADPKD).
Our ADPKD product candidate, RGLS8429, is being evaluated in a Phase 1b clinical trial. (NCT05521191)
About ADPKD.

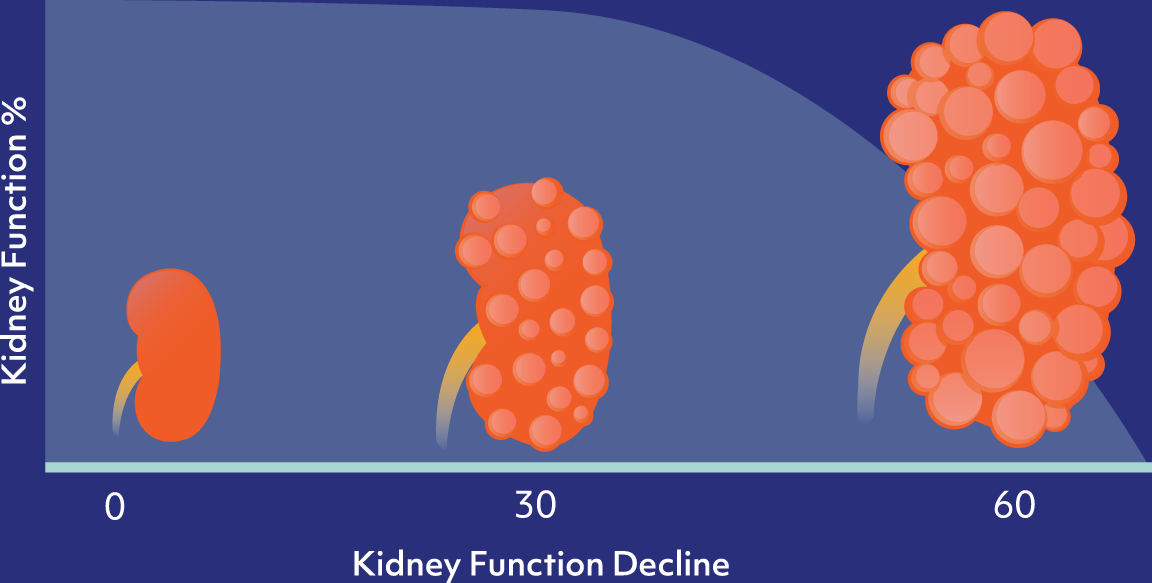
ADPKD is an orphan disease of high unmet need that effects approximately 160,000 diagnosed individuals in the U.S.
Total Patient Population
-
12M
Globally
-
500K
In the U.S.
of patients develop end stage renal disease by age 60 and require dialysis or transplantation

The only existing FDA-approved agent for ADPKD, tolvaptan, carries a boxed warning for potential fatal liver injury
CAUSES
ADPKD is caused by a mutation of either the Pkd1 or Pkd2 genes, which leads to the formation and proliferation of fluid-filled cysts in the kidneys, and ultimate loss of kidney function over time.